Integrating with MaldiAMRKit (MaldiSet)#
End-to-end workflow for users of the MaldiAMRKit ecosystem: start from a MaldiSet, delegate the batch-effect correction to MaldiBatchKit via MaldiSetAdapter, and keep MaldiAMRKit’s downstream machinery (.y, .filter(...), AMR evaluation) working on the corrected dataset.
Uses the MALDI-Kleb-AI dataset (Rocchi et al., 2026; Zenodo DOI 10.5281/zenodo.17405072); see notebook 01 for caching details. load_maldi_kleb_ai already builds a MaldiSet internally; we reach through ds.maldi_set to pass it to the adapter.
1. Load the MALDI-Kleb-AI MaldiSet#
[1]:
import sys, pathlib
sys.path.insert(0, str(pathlib.Path.cwd().parent))
from notebooks._demo import load_maldi_kleb_ai
demo = load_maldi_kleb_ai(antibiotic='Amikacin', verbose=True)
ds = demo.maldi_set
print(ds)
print('X shape:', ds.X.shape, '| y columns:', list(ds.y.columns))
ds.meta.head()
MaldiSet(n_spectra=743, species='all', antibiotics=['Amikacin'])
X shape: (741, 6000) | y columns: ['Amikacin']
[1]:
| Amikacin | Meropenem | Species | City | |
|---|---|---|---|---|
| ID | ||||
| FPG_2_9_spectrum | R | R | Klebsiella pneumoniae | Rome |
| FPG_2_15_spectrum | R | R | Klebsiella pneumoniae | Rome |
| FPG_2_47_spectrum | S | S | Klebsiella pneumoniae | Rome |
| FPG_2_55_spectrum | R | R | Klebsiella pneumoniae | Rome |
| FPG_2_57_spectrum | S | S | Klebsiella pneumoniae | Rome |
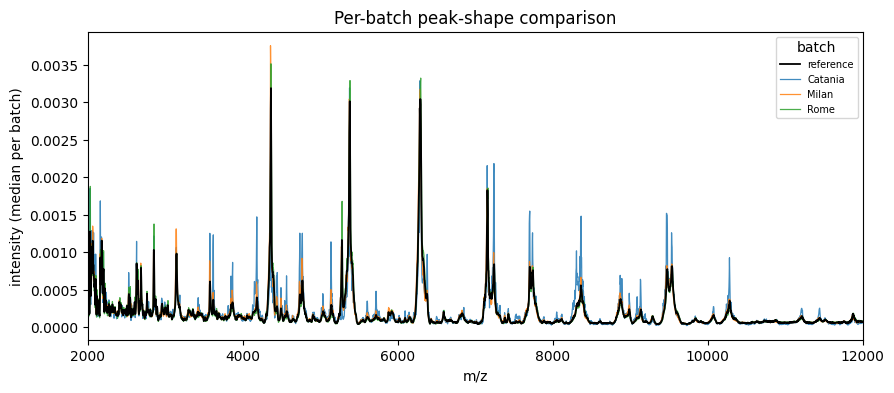
2. Inspect the cross-centre batch effect#
[2]:
import matplotlib.pyplot as plt
%matplotlib inline
from maldibatchkit.diagnostics import silhouette_batch, kbet
from maldibatchkit.viz import plot_peak_shift
batch = ds.meta['City'] # Rome / Milan / Catania
print(f'silhouette (before): {silhouette_batch(ds.X, batch):.3f}')
print(f'kBET acceptance (before): {kbet(ds.X, batch)["acceptance_rate"]:.3f}')
fig, ax = plot_peak_shift(batch, ds.X, mz_values=demo.mz)
ax.set_xlim(2000, 12000)
plt.show()
silhouette (before): 0.046
kBET acceptance (before): 0.239
3. Correct via MaldiSetAdapter#
[3]:
from maldibatchkit.integrations import MaldiSetAdapter
from maldibatchkit import SpeciesAwareComBat
adapter = MaldiSetAdapter(
batch_column='City',
species_column='Species',
quality_column=None, # SNR is stored on the DemoDataset, not on ds.meta
)
corrected_ds = adapter.correct(ds, SpeciesAwareComBat)
# Original dataset is untouched and the corrected MaldiSet keeps every property.
print('original X:', ds.X.shape, '| corrected X:', corrected_ds.X.shape)
print('AMR labels unchanged:', (ds.y.values == corrected_ds.y.values).all())
corrected_ds.meta.head()
original X: (741, 6000) | corrected X: (741, 6000)
AMR labels unchanged: True
[3]:
| Amikacin | Meropenem | Species | City | |
|---|---|---|---|---|
| 1-8317003599 | S | S | Klebsiella pneumoniae | Milan |
| 10-8320002130 | S | S | Klebsiella pneumoniae | Milan |
| 100-8660007296 | S | S | Klebsiella pneumoniae | Milan |
| 1004 | R | R | Klebsiella pneumoniae | Rome |
| 101-8140000209 | S | S | Klebsiella pneumoniae | Milan |
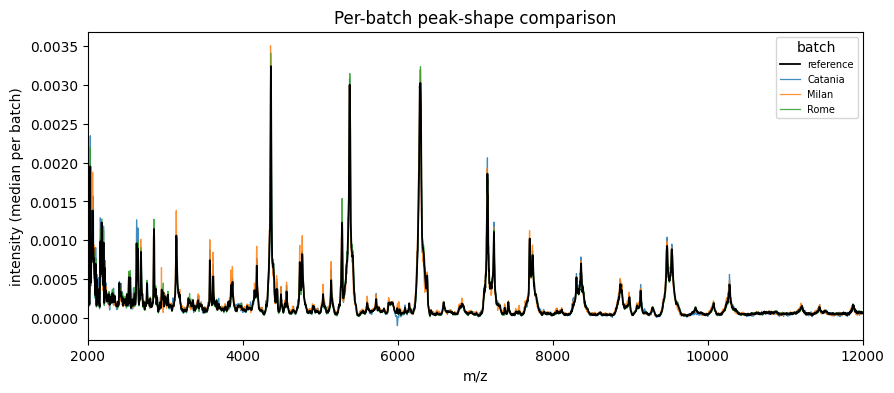
4. Verify the correction#
[4]:
print(f'silhouette (after): {silhouette_batch(corrected_ds.X, batch):.3f}')
print(f'kBET acceptance (after): {kbet(corrected_ds.X, batch)["acceptance_rate"]:.3f}')
fig, ax = plot_peak_shift(batch, corrected_ds.X, mz_values=demo.mz)
ax.set_xlim(2000, 12000)
plt.show()
silhouette (after): -0.001
kBET acceptance (after): 0.375
5. Swap in a quality-weighted corrector#
[5]:
from maldibatchkit import QualityWeightedComBat
# Supply the SNR column computed by load_maldi_kleb_ai directly via transformer_kwargs.
qw_ds = adapter.correct(
ds,
QualityWeightedComBat,
transformer_kwargs={'quality': demo.quality.loc[ds.X.index]},
)
print(f'silhouette (QW-ComBat): {silhouette_batch(qw_ds.X, batch):.3f}')
print(f'kBET acceptance (QW-ComBat): {kbet(qw_ds.X, batch)["acceptance_rate"]:.3f}')
silhouette (QW-ComBat): 0.011
kBET acceptance (QW-ComBat): 0.371
The adapter routes the Species column to SpeciesAwareComBat’s species argument automatically. Pass quality (SNR) through transformer_kwargs when it isn’t already a column on ds.meta; if your MaldiSet has one, set quality_column='<col_name>' and the adapter will route it for you.
6. Continue with MaldiAMRKit downstream tools#
[6]:
# The corrected MaldiSet behaves exactly like the original.
kp_mask = corrected_ds.meta['Species'] == 'Klebsiella pneumoniae'
print(f'K. pneumoniae samples: {kp_mask.sum()} / {len(corrected_ds.meta)}')
# AMR label prevalence after correction (labels are untouched)
corrected_ds.y.apply(lambda s: s.value_counts()).T
K. pneumoniae samples: 684 / 741
[6]:
| Amikacin | S | R |
|---|---|---|
| Amikacin | 372 | 369 |
Summary#
MaldiSetAdaptertakes aMaldiSetplus three column-name hints and returns a newMaldiSetwhose.Xyields the corrected matrix.Species-style metadata is routed to the transformer’s categorical covariate slot automatically.
The corrected dataset keeps working with every MaldiAMRKit feature:
.y,.meta,.filter(...), AMR evaluation, etc.